Change Language German
Overview of the revised IC3D Classification 2015
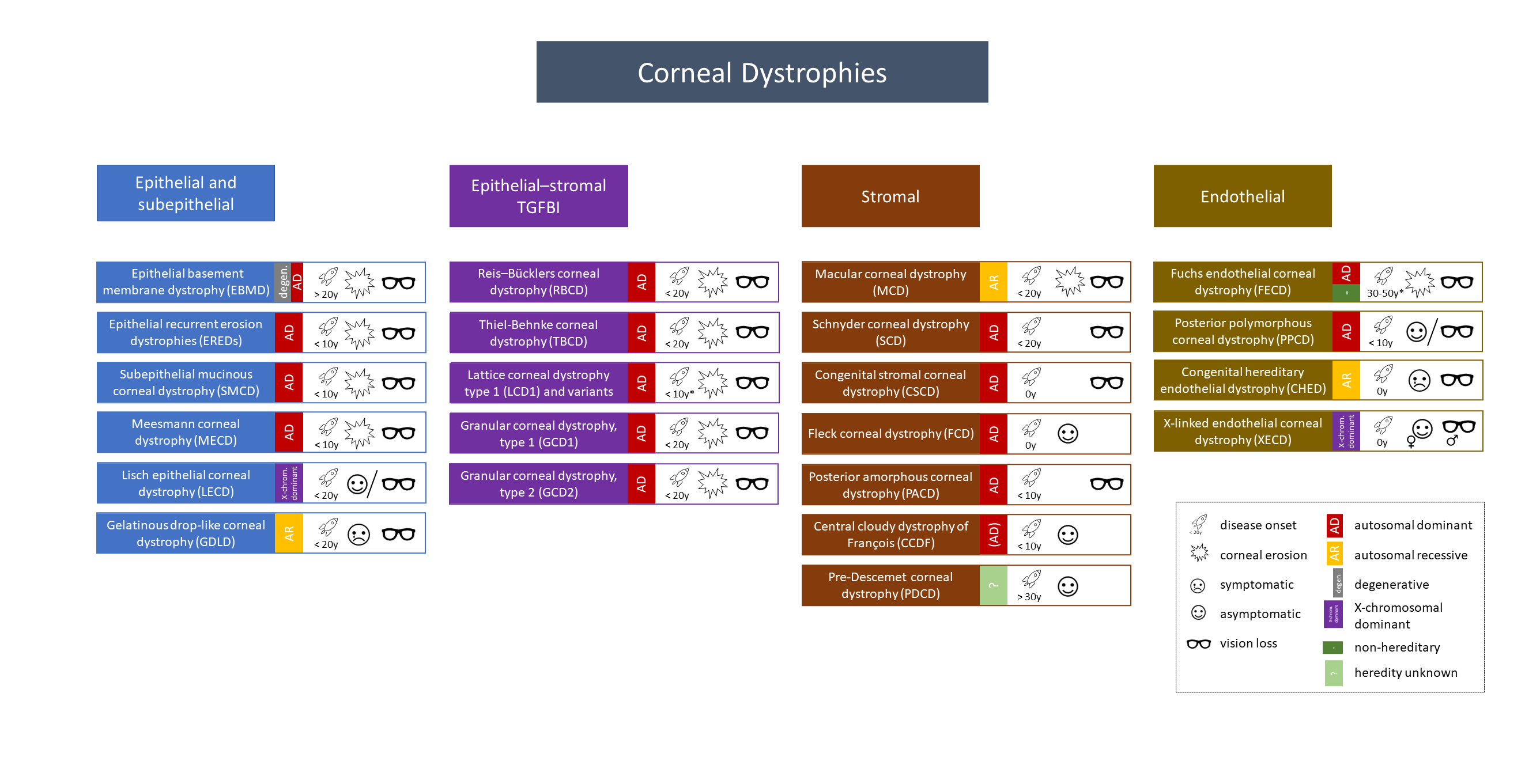
General
- Hereditary, bilateral, progressive corneal diseases
- Exceptions exist
- Family history important
- Examination: examine with dilated pupils, always examine the partner eye
- Categories
- Kategorien
- C1: Clinically and histologically well-defined dystrophy with identification of the gene and mutations
- C2: Clinically and histologically well-defined dystrophy with known chromosomal location and unknown gene identification
- C3: Clinically and histologically well-defined dystrophy without known chromosomal locus
- C4: Suspected new or previously documented dystrophy, not clear if distinct entity
- Mnemonic: Marilyn Monroe Always Gets Her Men in L. A. County
- Macular dystrophy – Mucopolysaccharide – Alcian blue
- Granular dystrophy – Hyaline materials – Masson trichrome
- Lattice dystrophy – Amyloid – Congo red
Epithelial and Subepithelial Dystrophies
Epithelial Basement Membrane Dystrophy (EBMD)
- see separate article: Map-Dot-Fingerprint-Dystrophy
Epithelial Recurrent Erosion Dystrophies (EREDs)
- Onset in the 1st decade (usually 4-6 years old), autosomal dominant, C4
- Variant: Dystrophia Smolandiensis C3
- Clinical presentation
- Recurrent epithelial corneal erosions in childhood, often without opacities
- Episodes with redness, photophobia, epiphora, eye pain, burning sensation, sensitive eyes over several years
- Induced by sunlight, dust, smoke, lack of sleep
- Adult patients with Smolandiensis variant may have central diffuse subepithelial opacities, sometimes keloid-like
- 25% undergo corneal transplantation around age 40 due to visual impairment
- Most patients have no pain attacks after the age of 50
Subepithelial Mucinous Corneal Dystrophy (SMCD)
- Onset in the 1st decade, autosomal dominant, C4 (likely belongs to EREDs)
- Allgemeines: Beginn in 1. Lebensjahrzehnt, autosomal dominant, C4 (gehört wahrscheinlich zu EREDs)
- Clinical presentation
- Recurrent epithelial corneal erosions in childhood
- Adults may have diffuse subepithelial opacities and haze
- Progressive visual loss in puberty
Meesmann Corneal Dystrophy (MECD)
- Onset in early childhood, autosomal dominant, C1
- Clinical presentation
- Often asymptomatic, mild visual impairments, occasional punctate epithelial erosions
- Diffuse gray opacities in direct illumination
- Multiple, solitary, round and clear epithelial vesicles in indirect illumination, sometimes extending to the limbus
- Slowly progressive
Lisch Epithelial Corneal Dystrophy (LECD)
- Onset in childhood, X-chromosomal dominant, C2
- Clinical presentation
- No difference between men and women!
- Asymptomatic or visual impairment with central changes, no erosions!
- Diffuse gray opacities in direct illumination
- Multiple, clustered, round and clear microcysts in indirect illumination
- Slow progression
Gelatinous Drop-like Corneal Dystrophy (GDLD)
- Onset in the 1st to 2nd decade, autosomal recessive, C1
- Clinical presentation
- Severe visual impairment, photophobia, irritation, redness, tearing
- Subepithelial changes (band keratopathy type), most common
- Multiple small mulberry-shaped nodules (mulberry type), delayed staining with fluorescein (permeable epithelium), often superficial vascularisation
- In later years, stromal opacification with larger nodular lesions (“kumquat type”)
- Progression, recurrence within a few years after superficial keratectomy or corneal transplantation
Epithelial-Stromal TGFBI Dystrophies
Reis-Bücklers Corneal Dystrophy (RBCD)
- Onset in childhood, autosomal dominant, C1, sometimes difficult to distinguish from TBCD
- Clinical presentation
- Frequently recurring erosions (more frequent and intense than TBCD), less severe in adulthood
- Slowly progressive visual loss
- Subepithelial, map-like opacities, can extend to the limbus and stroma
Thiel-Behnke Corneal Dystrophy (TBCD)
- Onset in childhood, autosomal dominant, C1/C2, sometimes challenging to distinguish from RBCD.
- Clinical presentation
- Recurrent erosions, milder than RBCD, may decrease over time
- Onset of visual impairment later than RBCD
- Subepithelial honeycomb-shaped opacities, can extend to the limbus and stroma, usually sparing the periphery
Lattice Corneal Dystrophy Type 1 (LCD1) and Variants
- Onset in the first decade of life, autosomal dominant, C1.
- Clinical presentation
- Recurrent erosions and visual impairment in the first two decades
- Paracentral, birefringent lattice lines, central progressive diffuse opacities; approximately 1mm peripheral zone remains clear
- Visual deterioration in the fourth decade, often leading to keratoplasty
- LCD Variants (Type IIIa, I/IIIa, IV, and polymorphic amyloidosis) occur later than Type 1
- LCD-Varianten (Typ IIIa, I/IIIa, IV und polymorphe Amyloidose) treten später auf als Typ 1
- Type IIIa: Thicker lines from limbus to limbus
- Type I/IIIa: Thinner lines
- Polymorphic amyloidosis: No lines
- Erosions in Type IIIa and I/IIIa, no erosions in Type IV and polymorphic amyloidosis
- Special Case: Formerly Gelsolin Type 2 (LCD2) = Meretoja Syndrome
- Generalised amyloidosis with corneal involvement; misleading term, not a corneal dystrophy
- Autosomal dominant, onset in the 3rd-4th decades, similar lattice lines as Lattice Dystrophy, but more peripheral
- Severe dermatochalasis and lagophthalmos due to cranial neuropathy with facial paralysis, increased risk of Primary Open-Angle Glaucoma (POAG), reduced corneal sensitivity, normal visual acuity until the sixth decade of life, increased dry eyes, and erosions in advanced age
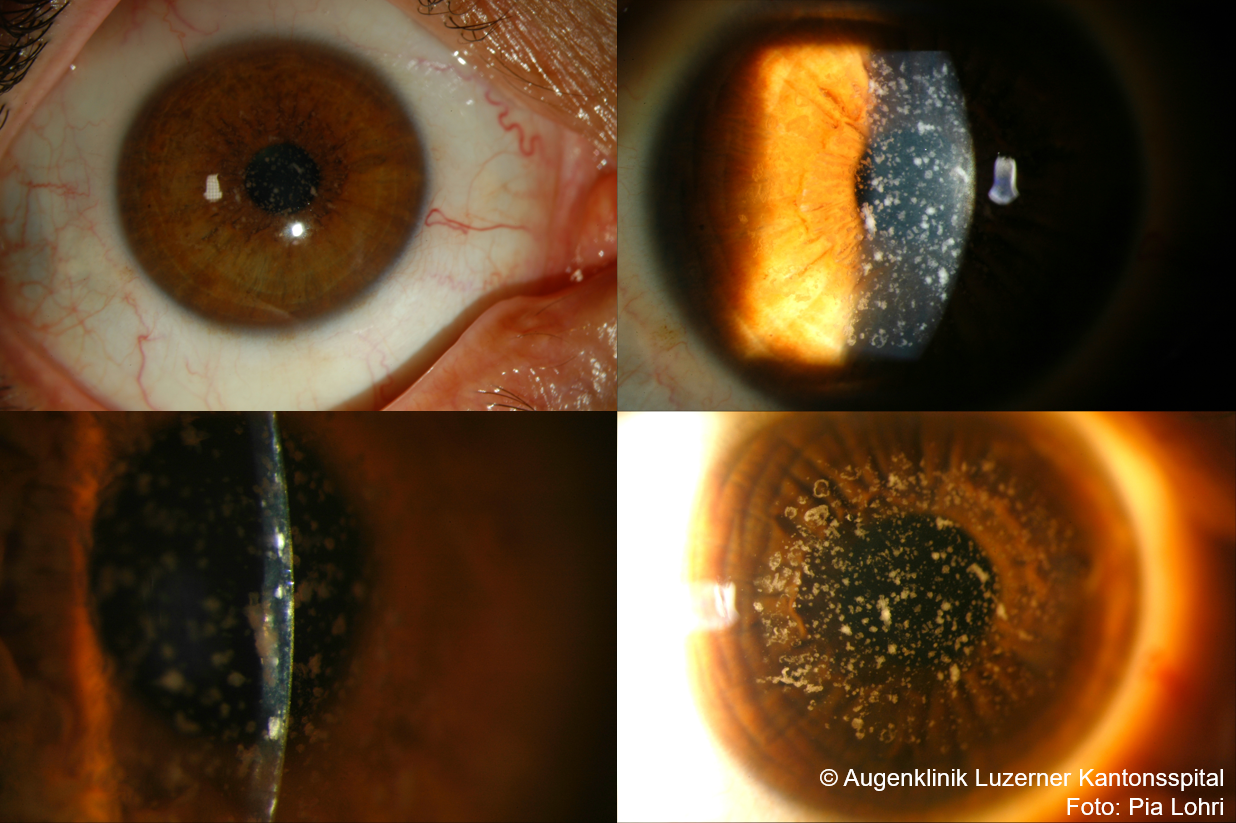
Granular Corneal Dystrophy Type 1 (GCD1)
- Onset in childhood, autosomal dominant, C1
- Clinical Presentation
- Frequent recurrent erosions, visual impairment with progressive confluent opacities in later years.
- Central well-defined particles/snowstorm in the anterior cornea, whitish under direct illumination; opacities do not extend to the limbus.
- Clear cornea between opacities (unlike macular corneal dystrophy)
{kind=link}
Granular Corneal Dystrophy Type 2 (GCD2)
- Onset in childhood, autosomal dominant, C1, formerly called Avellino Dystrophy
- Clinical presentation
- Initially fine white central subepithelial dot opacities, later rings and stars, often in finger-shaped patterns
- In the late phase, superficial, widened, breadcrumb-like, and partially translucent opacities extend into the anterior/mid stroma
- Clear cornea between opacities (unlike macular corneal dystrophy)
- Fewer opacities than Type 1
- Slow progression, visual impairment over time due to progressive central opacities, occasional erosions
Stromal Dystrophies
Macular Corneal Dystrophy (MCD)
- Onset in childhood, autosomal recessive, C1
- Clinical presentation
- Severe visual impairment between 10 and 30 years, recurrent erosions, reduced corneal sensitivity, photophobia
- Initially central, later fleck-like whitish opacities extending from the center to the periphery and Descemet membrane (unlike granular corneal dystrophy)
- Thinning of the central cornea is common
- Advanced stages may exhibit guttae with stromal edema
- Slow progression
Schnyder Corneal Dystrophy (SCD)
- Onset in childhood, autosomal dominant, C1, formerly Schnyder’s crystalline corneal dystrophy
- Diagnosis often made in the 2nd-3rd decades (more challenging in Type 2 without crystals)
- Clinical presentation
- Type 1: Central discoid/ring-shaped opacity, densely packed comma-shaped, multicolored subepithelial crystals
- Type 2: Central diffuse subepithelial discoid/ring-shaped opacity without crystals
- Both types associated with arcus lipoides
- Progressive development of diffuse stromal opacities
- Photopic vision significantly reduced despite typically good scotopic vision
- Slow progression, often requiring keratoplasty after 50 years due to photopic vision
Congenital Stromal Corneal Dystrophy (CSCD)
- Congenital, autosomal dominant, C1
- Clinical presentation
- Moderate to severe visual impairment
- Diffuse snowflake-like opacities throughout the corneal stroma, thickened cornea
- Non-progressive or slowly progressive
Fleck Corneal Dystrophy (FCD)
- Congenital, autosomal dominant, C1
- Clinical presentation
- Asymptomatic
- Solitary, scale-like, white stromal opacities in an otherwise clear cornea
- Can be asymmetric or unilateral
- Non-progressive
Posterior Amorphous Corneal Dystrophy (PACD)
- Onset in the 1st decade, autosomal dominant, C3 (possibly mesodermal dysgenesis)
- Clinical presentation
- Slightly reduced vision, usually > 0.5
- Diffuse opacity with transparent stromal breaks, especially in the posterior stromal area
- Associated with iris anomalies
- Non-progressive or slowly progressive
Central Cloudy Dystrophy of François (CCDF)
- Onset in the first decade of life, unknown etiology, autosomal dominant (occasionally described), C4.
- Clinical presentation
- Mostly asymptomatic
- Clinically identical to posterior Crocodile Chagrin
- Central cloudy, crocodile-skin-like opacities in the posterior stroma, with clear areas between them
- Non-progressive
Pre-Descemet Corneal Dystrophy (PDCD)
- Onset after the age of 30, unclear inheritance, C4
- Clinical presentation
- Asymptomatic, normal vision
- Point, line, ring, crystalline opacities in the posterior stromal area
- Similar findings to Ichthyosis
- No clinical difference between non-crystalline PDCD and Cornea farinata
- Non-progressive or slowly progressive (depending on the form)
Endothelial Dystrophies
Fuchs Endothelial Corneal Dystrophy (FECD)
- see separate article: Fuchs Endothelial Dystrophy
Posterior Polymorphous Corneal Dystrophy (PPCD)
- Onset in early childhood, autosomal dominant, C1-2
- Clinical presentation
- Often asymptomatic; significant vision loss possible with progression towards the stroma
- Endothelium behaves like epithelium
- Nodular, vesicular, band-shaped opacities, individually or in groups, in the posterior corneal area
- Typical railroad track-like changes
- Often no change in endothelial findings for years; slow progression possible
Congenital Hereditary Endothelial Dystrophy (CHED)
- Mostly congenital, autosomal recessive, C1.
- Note: Formerly divided into CHED1 and CHED2—obsolete, as CHED1 = PPCD
- Clinical presentation
- Significant vision loss, photophobia, mild epiphora
- Diffuse, milky corneal opacities, possibly combined with isolated gray spots
- Markedly thickened cornea (up to 2-3 times normal)
- Often accompanied by nystagmus
- Generally stable course
X-Linked Endothelial Corneal Dystrophy (XECD)
- Congenital, X-chromosomal dominant, C2
- In cases of congenital corneal changes, parents should be examined for endothelial changes (Differential Diagnosis: Congenital Glaucoma)
- Clinical presentation
- In Men: Vision loss, milky corneal opacities, moon-crater-like endothelial changes, secondary subepithelial band keratopathy, nystagmus possible, progressive
- In Women: Typically asymptomatic, moon-crater-like endothelial changes, usually non-progressive
Sources
- Cornea-Society – Publications: IC3D Classifikation in several languages
- Weiss JS, Møller HU, Aldave AJ, Seitz B, Bredrup C, Kivelä T, Munier FL, Rapuano CJ, Nischal KK, Kim EK, Sutphin J, Busin M, Labbé A, Kenyon KR, Kinoshita S, Lisch W. IC3D classification of corneal dystrophies–edition 2. Cornea. 2015 Feb;34(2):117-59. doi: 10.1097/ICO.0000000000000307. Erratum in: Cornea. 2015 Oct;34(10):e32. PMID: 25564336.
- Lisch, Walter & Seitz, Berthold. (2011). Neue internationale Klassifikation der Hornhautdystrophien. Der Ophthalmologe. 108. 883-897. 10.1007/s00347-011-2388-8.
- Seitz B, Lisch W, Weiss J, Die revidierte neueste IC3D-Klassifikation der Hornhautdystrophien; Klin Monatsbl Augenheilkd 2015; 232: 283-294, DOI 10.1055/s-0041-100774